
PhD student Joseph Hawksworth describes his work developing tools to genetically engineer red blood cells
My last three years have been spent engineering red blood cells, working towards the ultimate goal of production of customised red blood cells for therapeutic uses. I’m a final year PhD student studying Synthetic Biology at the University of Bristol and I work in Dr Ashley Toye’s group which specialises in cultured blood in the laboratory. There are many definitions of Synthetic biology but common to them all is an emphasis on the design and engineering of biological systems for useful purposes. In the spirit of this, we aim to produce ‘designer’ red blood cells (RBCs) to help overcome numerous challenges, for example replicating disease states for study, improving properties of lab grown blood, or even utilising RBCs as drug delivery vehicles. This month my work was published in EMBO Molecular Medicine (DOI: 10.15252/emmm.201708454). Here I’ll give some background to the project and describe the challenges, failures, and successes along the way!
In order to produce designer RBCs, we needed to develop a reliable way of editing the genome. Fortunately, I joined the lab at an opportune moment as two exciting technologies were emerging: CRISPR-Cas9 gene editing and a new RBC precursor cell line called BEL-A (Bristol Erythroid Line – Adult). CRISPR-Cas9 technology was being hailed as a revolutionary new approach to gene editing, with many laboratories reporting successful edits in a variety of cell types. CRISPR-Cas9 consists of two components: Cas9, a protein which cuts DNA, and a guide RNA which binds to Cas9 and directs it to the target DNA site. Once DNA has been cut it can be repaired, either through faulty DNA repair, preventing gene function (knockout), or through repair using a third component, template DNA, which enables very precise edits of the DNA sequence. Whilst CRISPR gene editing was gaining in popularity, my co-supervisor, Dr Jan Frayne, was developing the BEL-A cell line which offered an endless supply of RBC precursor cells. The BEL-A cells offered an excellent testbed for CRISPR based editingand the goal of my PhD was to bring together these two nascent technologies.
Encouraged by CRISPR success stories from laboratories worldwide, we started with an ambitious attempt to insert a large piece of DNA into the genome. Unfortunately this was unsuccessful, so we opted for a simpler approach, creating gene knockouts. We tested a variety of ways for getting the CRISPR components into the BEL-A cells and found a lentiviral approach to be most efficient. Initially, genes associated with membrane proteins were targeted, such as glycophorin A, as these were easiest to detect presence/absence from the cell surface. This time the editing worked! We were excited at the implications; most blood groups are determined by membrane proteins (or sugars attached to these proteins), therefore the removal of membrane proteins potentially enables us to make RBCs with increased transfusion compatibility.
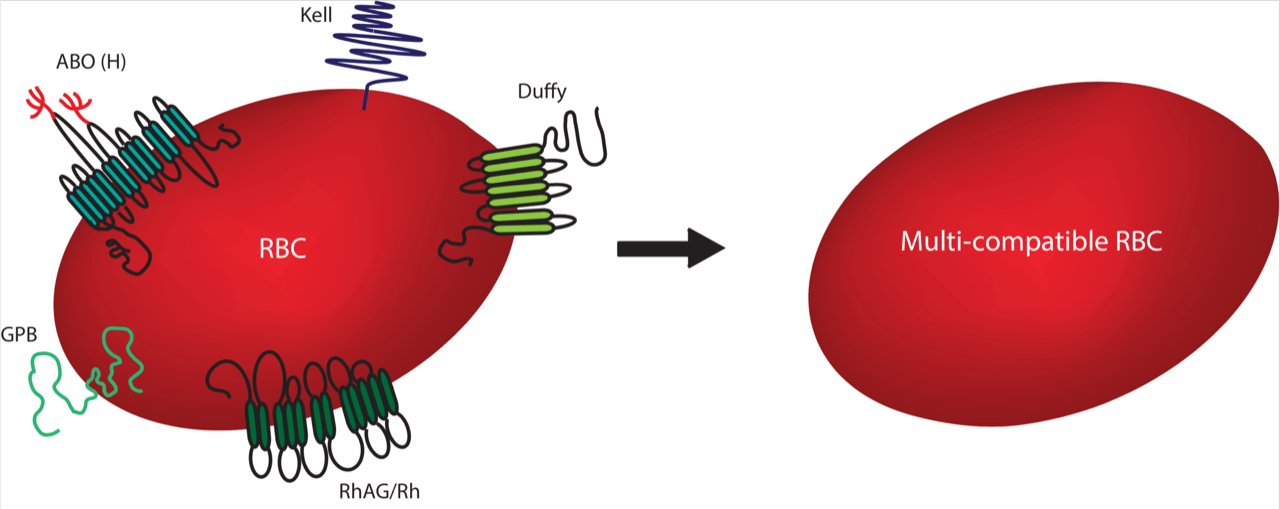
It can be extremely difficult to source blood for transfusions of people with very rare blood types. Likewise, people with blood diseases, such as sickle cell anemia and thalassemia, often require repeated blood transfusions with difficult-to-source blood which has been carefully matched to minor blood group antigens in order to avoid adverse transfusion reactions. We wanted to target proteins which, if removed, would theoretically serve the broadest possible transfusion requirement. To identify a ‘hit list’ of proteins for removal, we collaborated with Dr Fiona Regan (NHSBT Hammersmith) who conducted a 15-month NHSBT survey to identify which antigens most commonly caused difficulty in matching blood for transfusions.The blood group proteinsRh, Kell, Duffy and GPB, as well as the ABO group (a carbohydrate), were identified as key targets. Using CRISPR-Cas9 we were able to generate BEL-A lines deficient for each of these targets and confirmed that the cells were still capable of developing into reticulocytes (young RBCs).
The next stage was to combine all of the above blood group knockouts in a single cell to produce a cell with a theoretically enhanced transfusion compatibility. We completed several rounds of gene editing until we had knocked out all targets. One multiple knockout cell line was selected and we showed that it was possible to make reticulocytes which looked identical and had very similar properties to the original cells. The whole genome was sequenced and we found no extra mutations caused by the CRISPR-Cas9 edits, other than the ones we expected to observe. This is the first RBC precursor line developed which is absent for all of these blood group proteins and it was intriguing to see that these absences could be tolerated in reticulocytes derived from this cell line.These cells could theoretically serve 48 out of the 56 cases identified in the initial survey,representing a significant step towards the ability to tailor blood groups to meet the needs of patients!
Despite the significant progress we’ve made in developing engineered RBCs, there remain a number of challenges before translating our work to a clinical scenario. The work has been completed on a laboratory scale, and the numbers of cells we have been working with would require significant scale up. Similarly, the current costs of cell culturing are prohibitively expensive for therapeutic use. Finally, the cell line is currently not approved for use in humans, and overcoming this would require the production of clinical-grade cells produced according to ‘good manufacturing practice’ requirements. However, this is important proof-of-concept work which demonstrates the potential for designer RBCs developed for therapeutic use in humans. The first recipients of such cells would likely be people who are difficult, if not impossible, to match blood for transfusion requirements at present. The next stage of this work for the Toye lab is developing more advanced CRISPR methods allowing greater control over the genome and this is what I’m currently working on.
On a personal note, I feel extremely lucky to publish the first paper of my PhD on such an exciting and fast-moving project. I am grateful to the publication’s co-authors, in particular Tim Satchwell who I worked closely with, and all members of the Toye laboratory, for their insight, support and collaboration. Now to write my thesis!